Summary information and primary citation
- PDB-id
- 1brn; SNAP-derived features in text and JSON formats;
DNAproDB
- Class
- hydrolase-DNA
- Method
- X-ray (1.76 Å)
- Summary
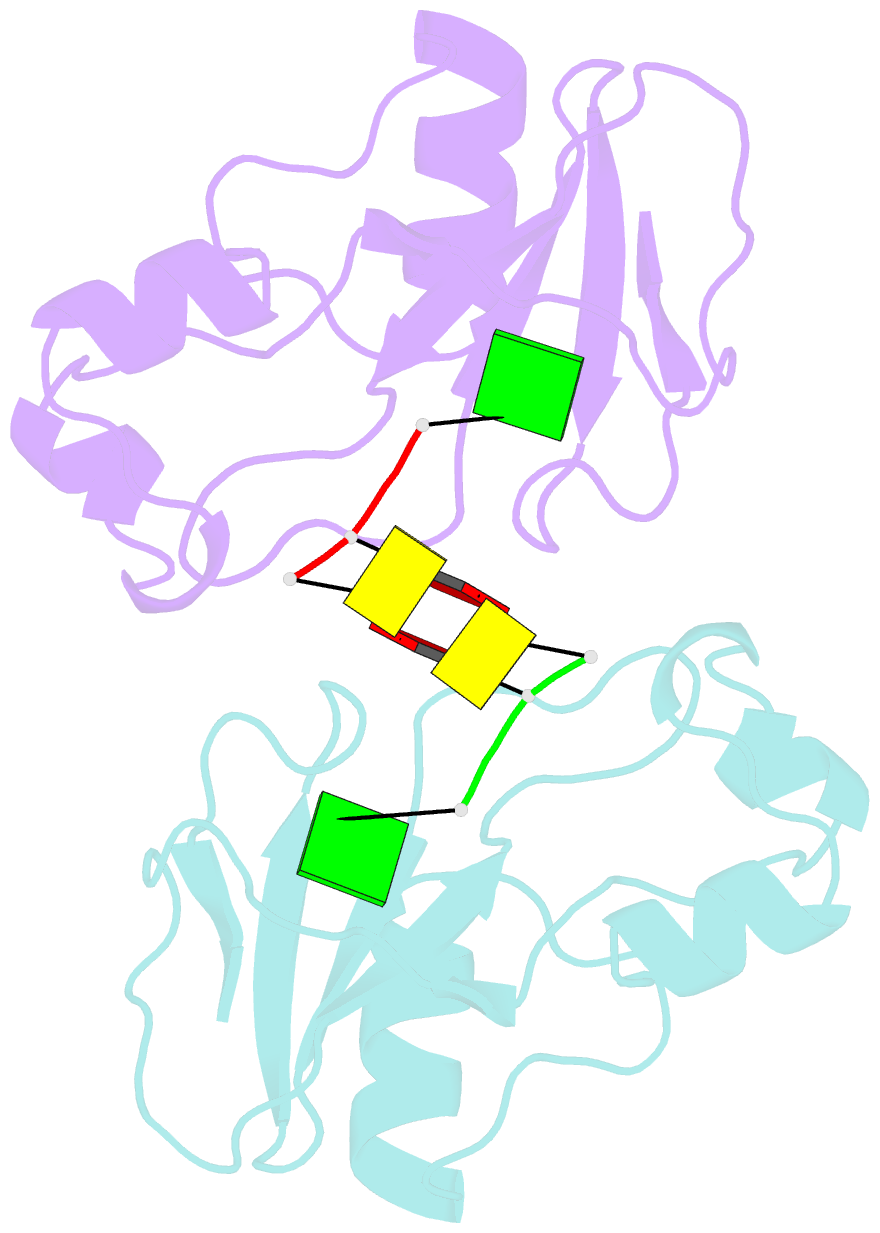
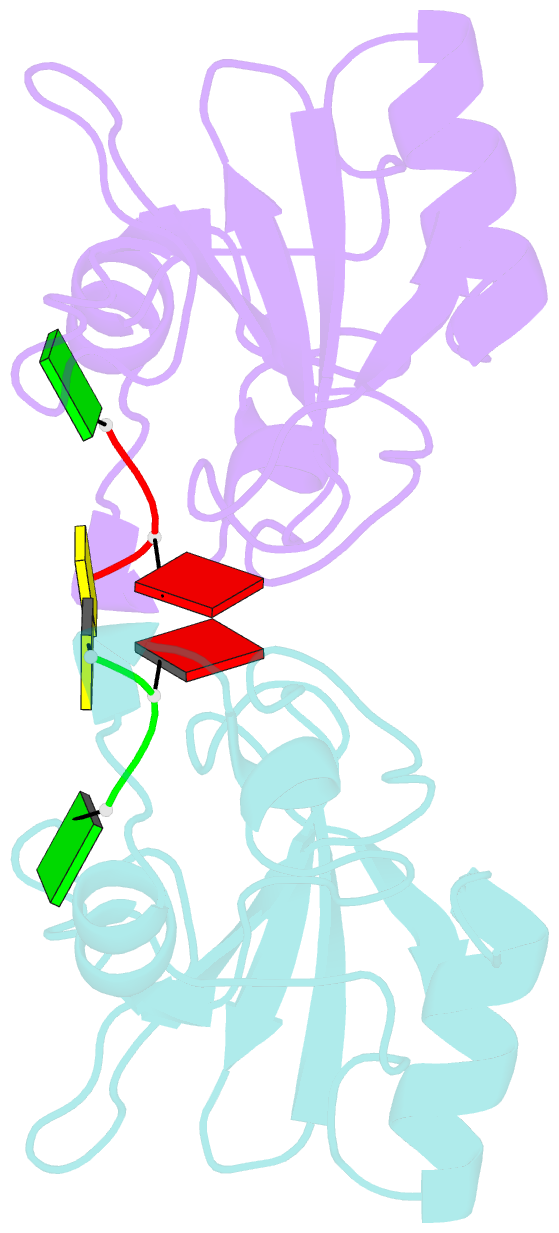
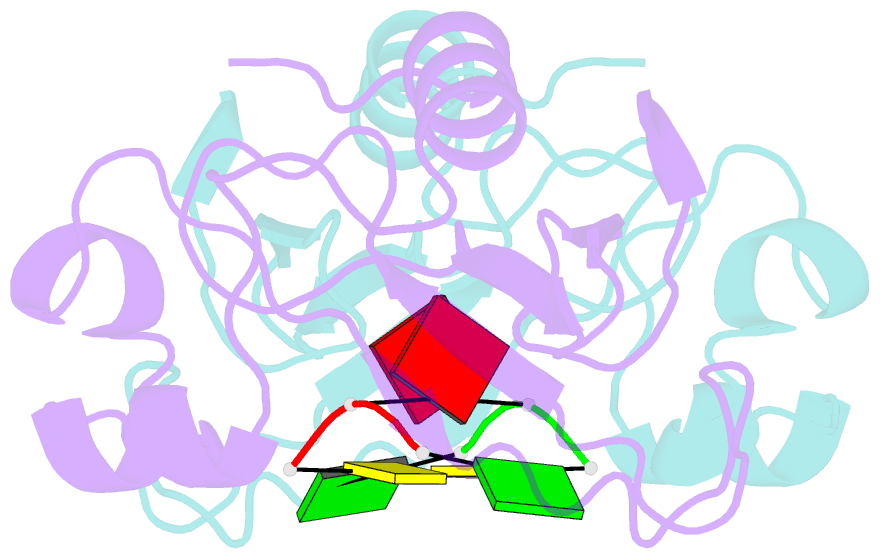
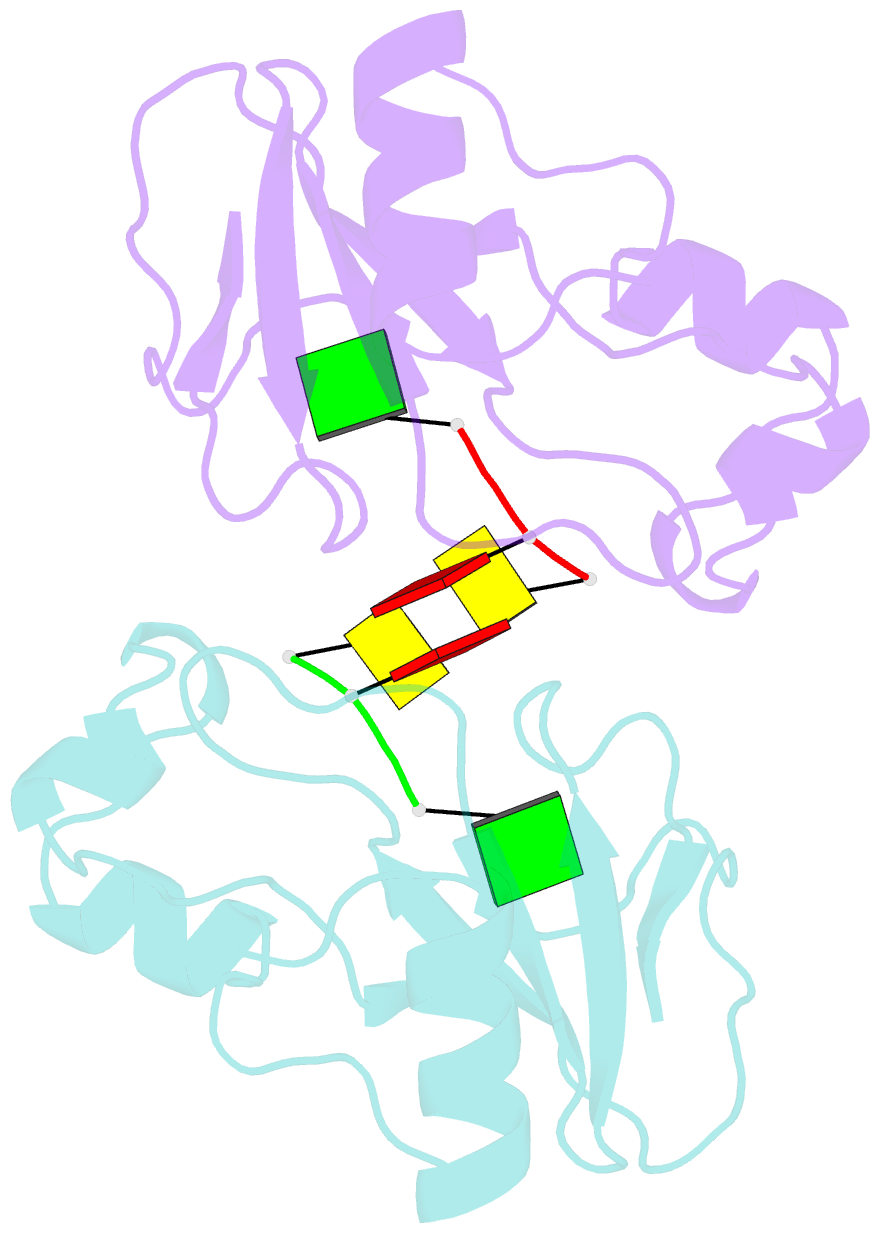
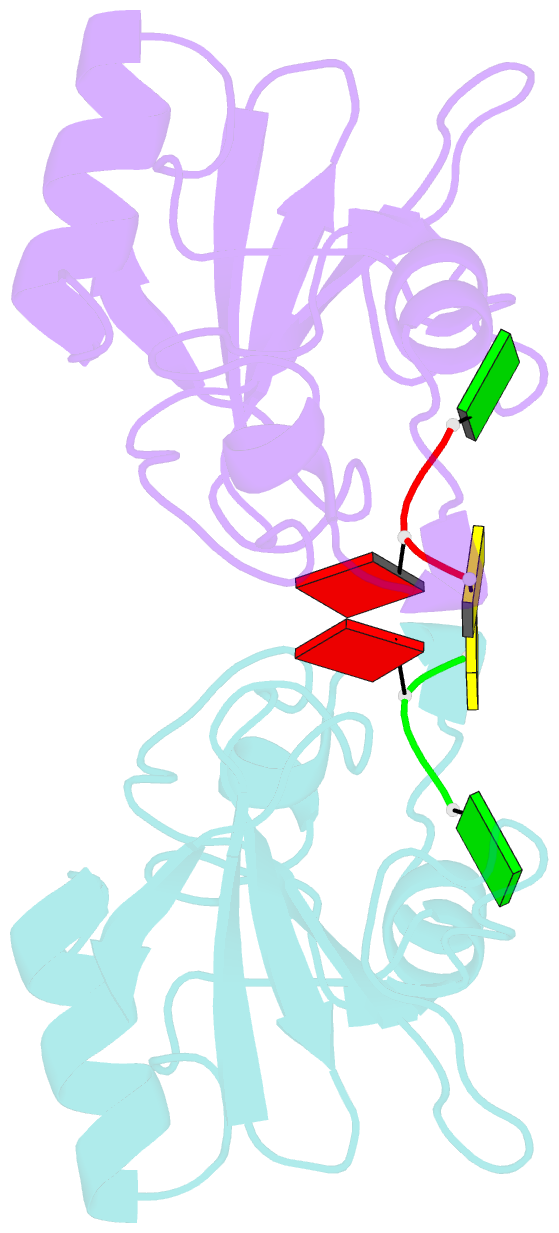
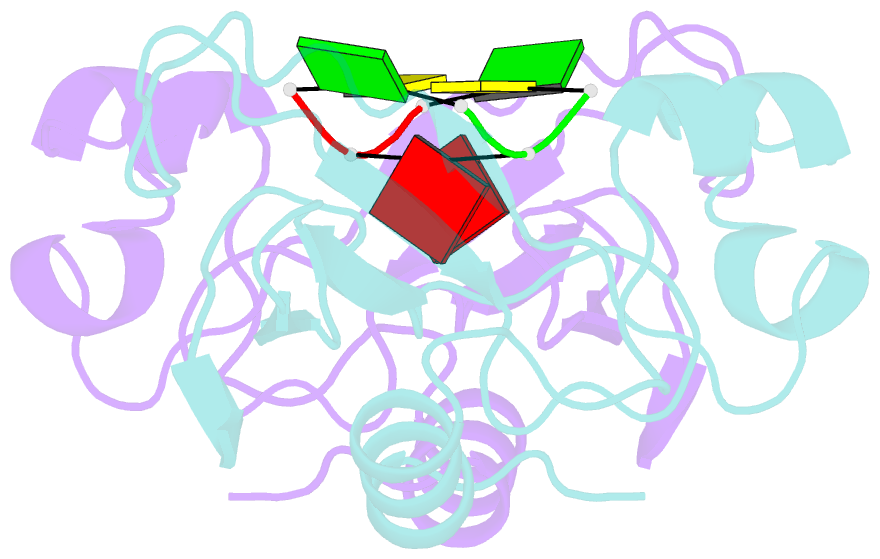
- Subsite binding in an rnase: structure of a barnase-tetranucleotide complex at 1.76 angstroms resolution
- Reference
- Buckle AM, Fersht AR (1994): "Subsite binding in an RNase: structure of a barnase-tetranucleotide complex at 1.76-A resolution." Biochemistry, 33, 1644-1653. doi: 10.1021/bi00173a005.
- Abstract
- A set of subsites in barnase has been proposed from kinetic studies. A specific substrate analog, the tetradeoxynucleotide, CGAC, has been designed from this information. We report the crystal structure of its complex with barnase at 1.76-A resolution. The structure was solved by molecular replacement from a model of free barnase and refined to a crystallographic R factor of 19.0%. The stoichiometry of the asymmetric unit dimeric complex is [barnase:d(CGAC)]2, with 2-fold noncrystallographic symmetry. Each barnase molecule binds one oligonucleotide whereby the recognition site is occupied by guanine, and all three phosphate groups of the nucleotide make electrostatic interactions with basic residues in a strongly electropositive region at the bottom of the active site. The active-site His 102 packs against the adenine base of the nucleotide in an almost identical manner to the guanine base in the barnase-d(GpC) complex and defines a possible subsite in the Michaelis complex. The overall protein structure is unchanged on forming the complex with d(CGAC), but there are small differences in the active site and in crystal packing regions. The protein coordinates will be useful for theoretical calculations since some disorder induced by packing constraints in the crystals of the free enzyme are absent in the crystals of the complex. The interface of the dimer is formed by a His 102-adenine-adenine-His 102 face-to-face ring stack directly on the 2-fold axis. The edge of the adenine-adenine stack packs closely onto the face of a 3'-cytosine-3'-cytosine interaction, which has a "base-pair"-like conformation but too great a separation of the bases to form hydrogen bonds. This unusual arrangement is the major stabilizing interaction within the dimeric complex, since there are no direct protein-protein interactions. Using the structure of the complex as a starting point for model building, the nature of the enzyme-substrate and enzyme-transition state complexes is investigated.