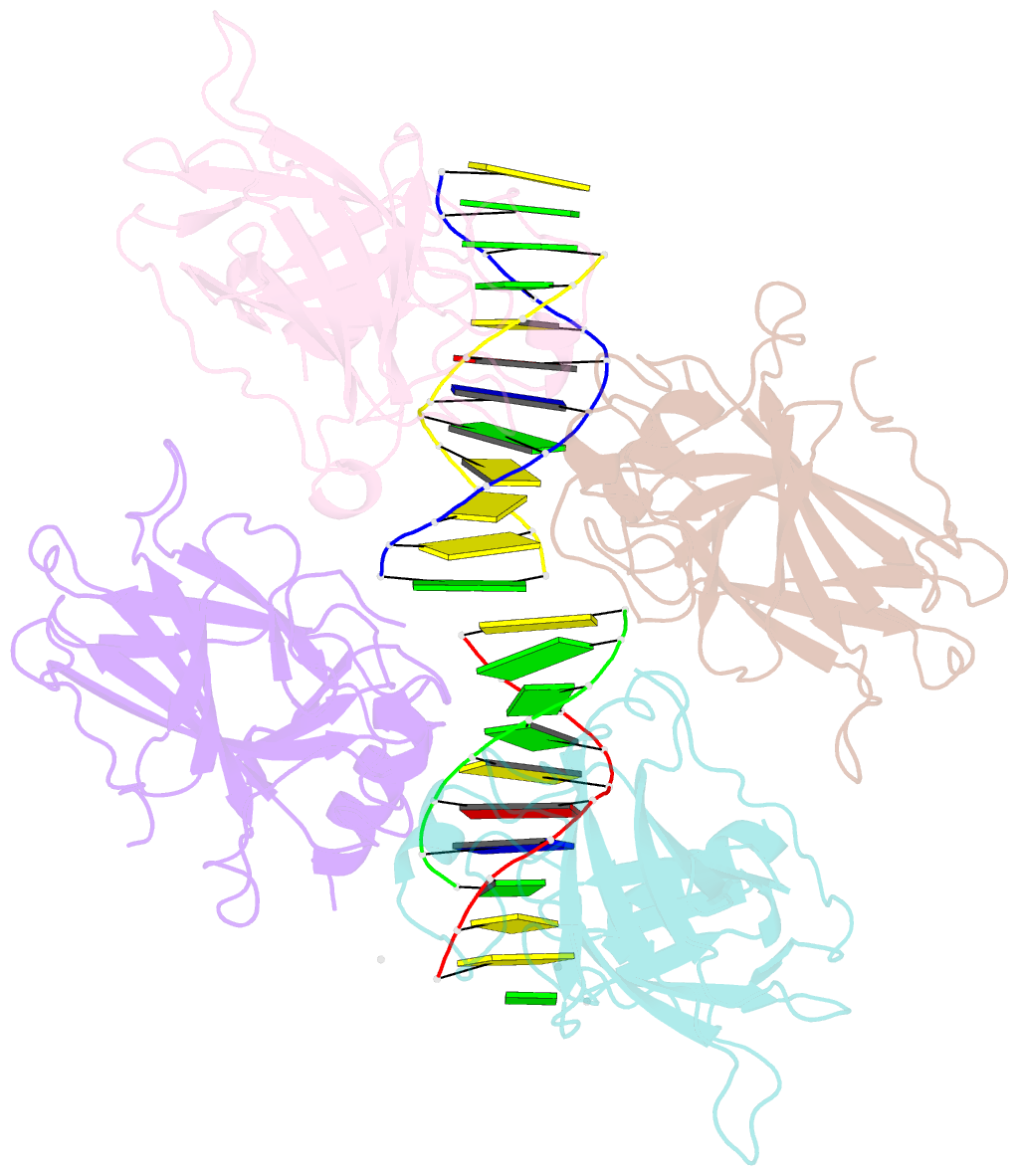
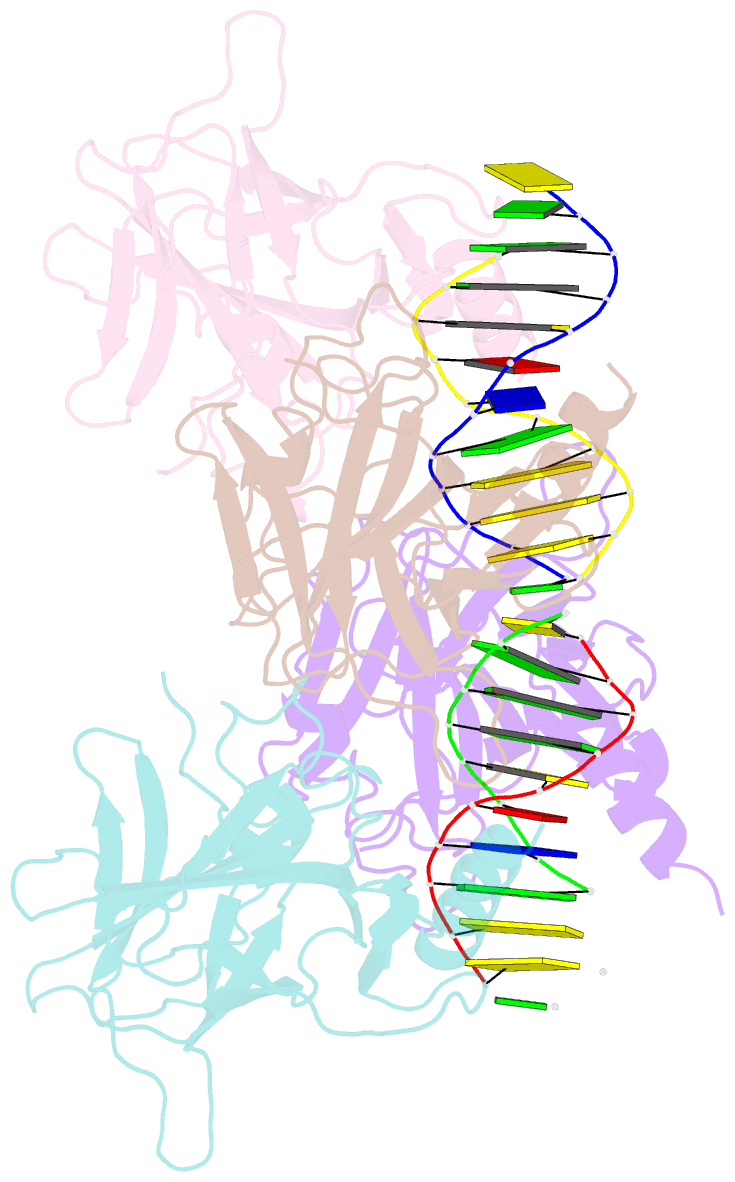
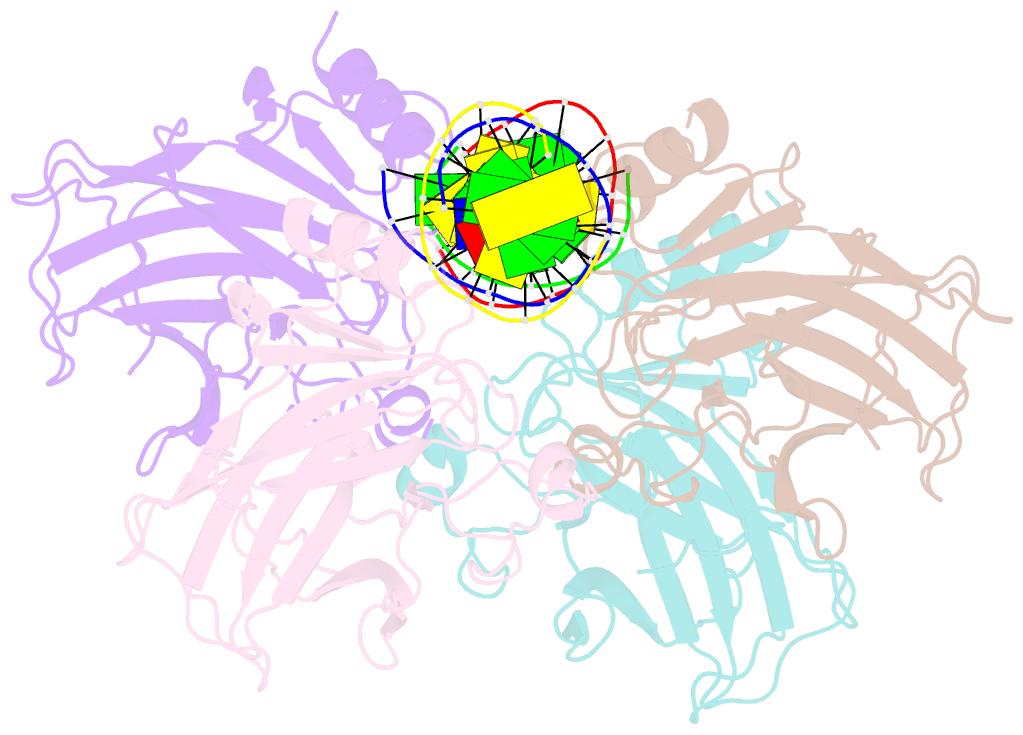
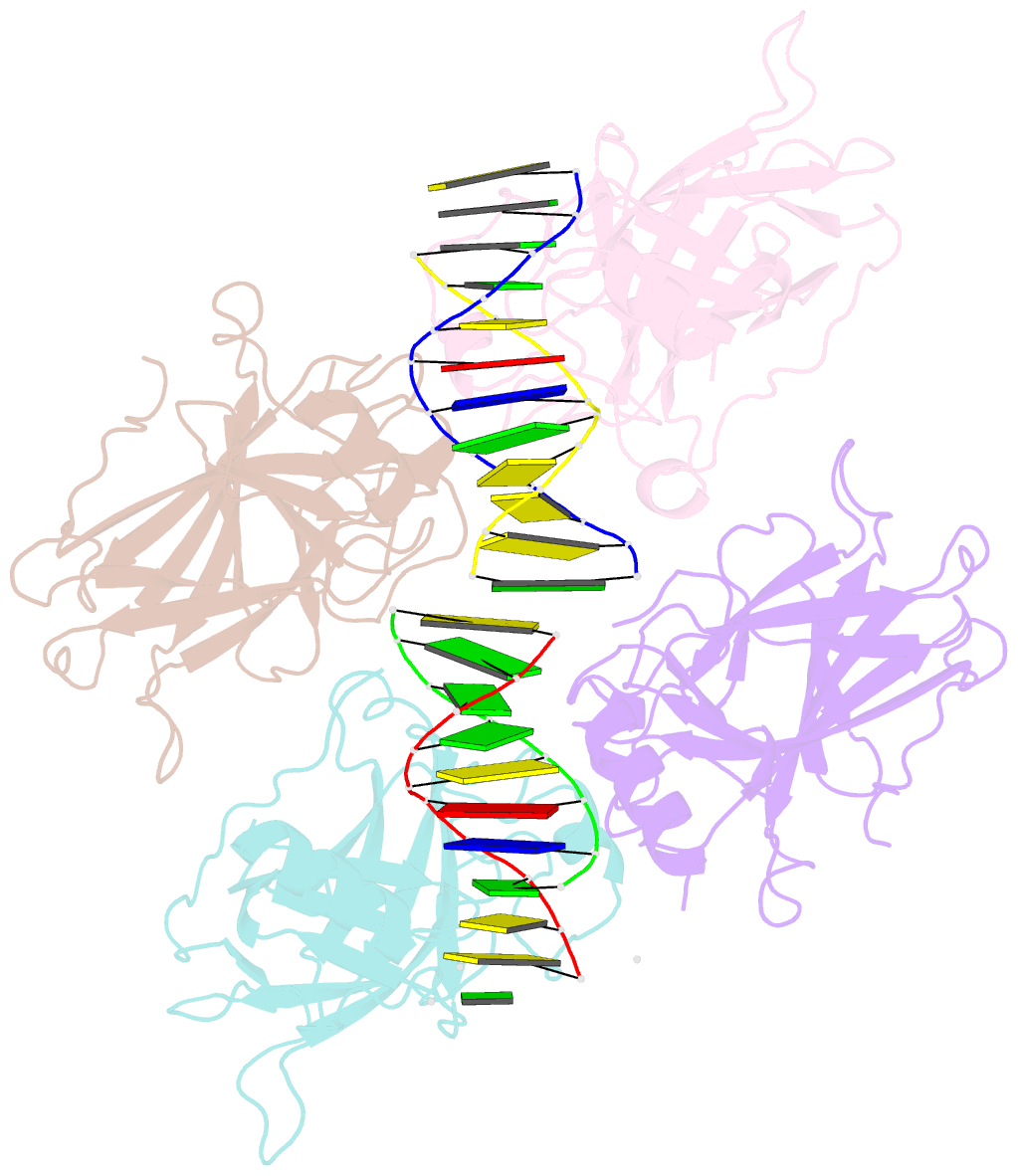
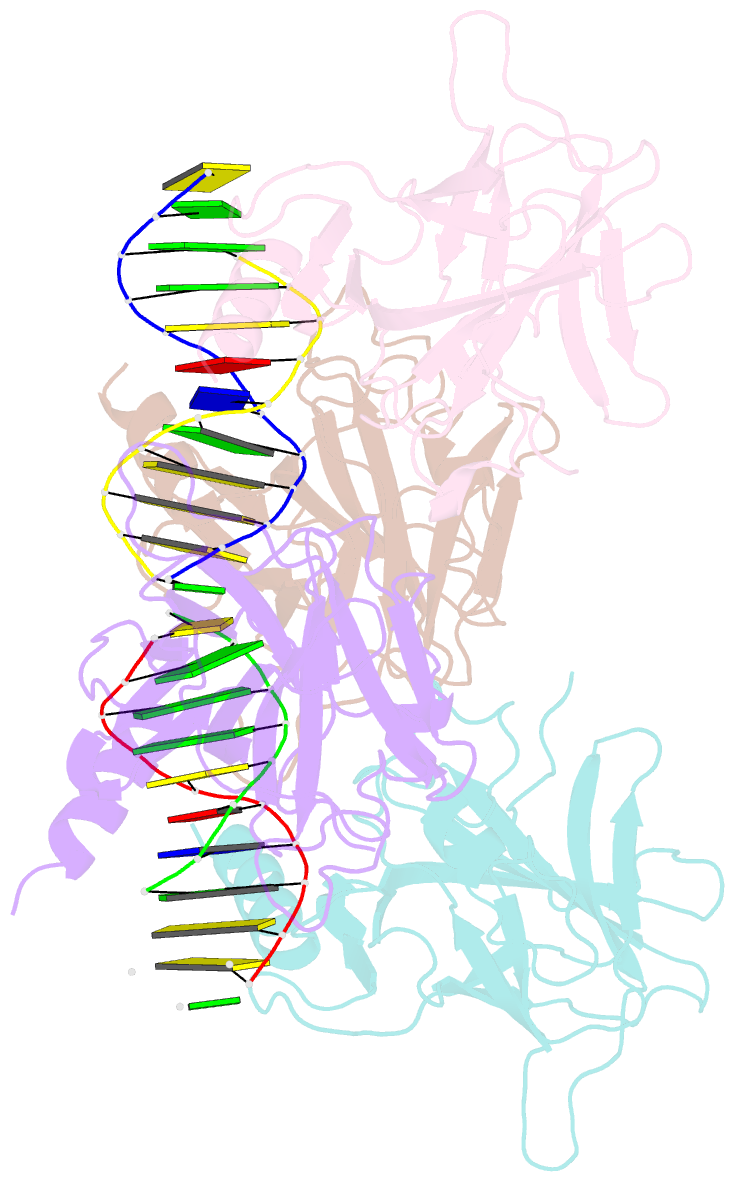
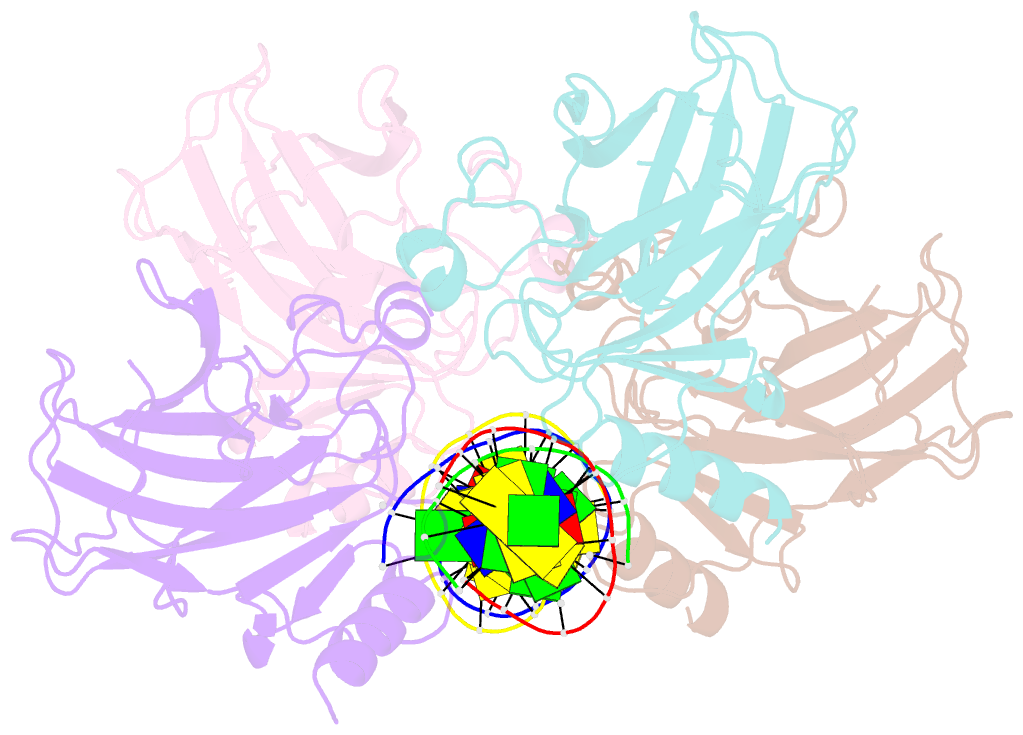
Summary information and primary citation
- PDB-id
- 3d0a; SNAP-derived features in text and JSON formats;
DNAproDB
- Class
- transcription-DNA
- Method
- X-ray (1.8 Å)
- Summary
- Human p53 core domain with hot spot mutation r249s and second site suppressor mutation h168r in sequence-specific complex with DNA
- Reference
- Suad O, Rozenberg H, Brosh R, Diskin-Posner Y, Kessler N, Shimon LJW, Frolow F, Liran A, Rotter V, Shakked Z (2009): "Structural basis of restoring sequence-specific DNA binding and transactivation to mutant p53 by suppressor mutations." J.Mol.Biol., 385, 249-265. doi: 10.1016/j.jmb.2008.10.063.
- Abstract
- The tumor suppressor protein p53 is mutated in more than 50% of invasive cancers. About 30% of the mutations are found in six major "hot spot" codons located in its DNA binding core domain. To gain structural insight into the deleterious effects of such mutations and their rescue by suppressor mutations, we determined the crystal structures of the p53 core domain incorporating the hot spot mutation R249S, the core domain incorporating R249S and a second-site suppressor mutation H168R (referred to as the double mutant R249S/H168R) and its sequence-specific complex with DNA and of the triple mutant R249S/H168R/T123A. The structural studies were accompanied by transactivation and apoptosis experiments. The crystal structures show that the region at the vicinity of the mutation site in the R249S mutant displays a range of conformations [wild-type (wt) and several mutant-type conformations] due to the loss of stabilizing interactions mediated by R249 in the wt protein. As a consequence, the protein surface that is critical to the formation of functional p53-DNA complexes, through protein-protein and protein-DNA interactions, is largely distorted in the mutant conformations, thus explaining the protein's "loss of function" as a transcription factor. The structure of this region is restored in both R249S/H168R and R249S/H168R/T123A and is further stabilized in the complex of R249S/H168R with DNA. Our functional data show that the introduction of H168R as a second-site suppressor mutation partially restores the transactivation capacity of the protein and that this effect is further amplified by the addition of a third-site mutation T123A. These findings together with previously reported data on wt and mutant p53 provide a structural framework for understanding p53 dysfunction as a result of oncogenic mutations and its rescue by suppressor mutations and for a potential drug design aimed at restoring wt activity to aberrant p53 proteins.